Code
library(qs2)
library(Seurat)
library(patchwork)
library(tidyverse)
library(DESeq2) # needed by FindAllMarkers(test.use = "DESeq2") in eval:false chunks
library(jsonlite)Identification of Astrocyte subpopulations after subsetting and reclustering
library(qs2)
library(Seurat)
library(patchwork)
library(tidyverse)
library(DESeq2) # needed by FindAllMarkers(test.use = "DESeq2") in eval:false chunks
library(jsonlite)::: {.cell}
# genes to remove #########################################################
# Load gene list and extract CELL SPECIFIC markers (excluding ASTROCYTE)
gene_list <- read_json("results/Xenium_Final_Gene_List_Updated_20260220_corrected.json")
cell_specific <- gene_list[["CELL SPECIFIC"]]
cell_specific[["ASTROCYTE"]] <- NULL
# One row per gene with cell_type label
markers <- map_dfr(names(cell_specific), function(ct) {
tibble(cell_type = ct, gene = unlist(cell_specific[[ct]]))
}) |>
pull(gene)
### genes manually selected by Akhil ###
akhil <- c(
"Tmem119", "Aif1", "Trem2", "P2ry12", "Ermn", "Cldn11", "Mog",
"Ccdc153", "Dnah11", "Tmem212", "Pdgfra", "Tnr", "Mgp", "Bgn",
"Slc47a1", "Acta2", "Tagln", "Vtn", "Kcnj8", "Atp13a5", "Flt1",
"Emcn", "Cldn5", "Car12", "Ttr", "Col1a2", "Lum", "Dcx", "Pax6",
"Mgl2", "Mrc1", "Cd3d", "Gzmb", "Pdcd1", "Klrb1c", "Tpsab1",
"Tpsb2", "Fcer1a", "Plac8", "Msr1", "Ly6g", "Camp", "Cd209a",
"Clec10a", "Cd247", "Snap25", "Grin2a", "Grin2b", "Cux2", "Otof",
"Stard8", "Lypd1", "Lrg1", "Adamts2", "Macc1", "Rapgef3", "Rorb",
"Tcap", "Hsd11b1", "Rspo1", "Whrn", "Tunar", "Osr1", "Oprk1",
"Pou3f1", "Tshz2", "Tox2", "Syt6", "Trh", "Vip", "Calb2", "Pthlh",
"Crh", "Gad1", "Serpinf1", "Cnr1", "Lhx6", "Slc32a1", "Slc17a7",
"Fezf2", "Htr2c", "Dpp4", "Slc17a6", "Siglech", "Clec7a", "Lyz2",
"Cd14", "Cst7", "Pf4", "Nkg7", "Cx3cr1", "Itgax", "Cd68", "Mbp",
"Mag", "Pllp", "Lamp5", "Nts", "Tac1", "Sncg", "Sst", "Calb1",
"Ndnf")
# setdiff(akhil, markers):::
Removed cell markers Tmem119, Aif1, Trem2, P2ry12, Ermn, Cldn11, Mog, Ccdc153, Dnah11, Tmem212, Pdgfra, Tnr, Mgp, Bgn, Slc47a1, Acta2, Tagln, Vtn, Kcnj8, Atp13a5, Flt1, Emcn, Cldn5, Car12, Ttr, Col1a2, Lum, Dcx, Pax6, Mgl2, Mrc1, Cd3d, Gzmb, Pdcd1, Klrb1c, Tpsab1, Tpsb2, Fcer1a, Plac8, Msr1, Ly6g, Camp, Cd209a, Clec10a, Cd247, Snap25, Grin2a, Grin2b, Cux2, Otof, Stard8, Lypd1, Lrg1, Adamts2, Macc1, Rapgef3, Rorb, Tcap, Hsd11b1, Rspo1, Whrn, Tunar, Osr1, Oprk1, Pou3f1, Tshz2, Tox2, Syt6, Trh, Vip, Calb2, Pthlh, Crh, Gad1, Serpinf1, Cnr1, Lhx6, Slc32a1, Slc17a7, Fezf2, Htr2c, Dpp4, Slc17a6 and Akhil´s hand-picked genes Siglech, Clec7a, Lyz2, Cd14, Cst7, Pf4, Nkg7, Cx3cr1, Itgax, Cd68, Mbp, Mag, Pllp, Lamp5, Nts, Tac1, Sncg, Sst, Calb1, Ndnf
# Load seurat with subsetted astrocytes object
astro_subset <- qs_read("seurat_objects/20260305-astro_0.3.qs2")
## NOT USED ####################################################
###### cells in cluster 0 expressing Cxcl10 ######
cxcl10_high_c0_cells <- WhichCells(
astro_subset,
idents = "0",
expression = Cxcl10 >= 0)
astro_subset <- subset(astro_subset, cells = setdiff(Cells(astro_subset),
astro0_Cxcl10))
############################################################
genes_to_remove <- akhil
all_features <- Features(astro_subset)
keep_genes <- all_features[!all_features %in% genes_to_remove]
astro_cleaned <- subset(astro_subset, subset = seurat_clusters!=6)
astro_cleaned <- subset(astro_cleaned, features = keep_genes)
# Keep only the assay needed for fresh SCTransform
DefaultAssay(astro_cleaned) <- "Xenium"
astro0 <- DietSeurat(astro_cleaned, assays = "Xenium", dimreducs = NULL, graphs = NULL)
# Drop Xenium image/FOV segmentation data; otherwise merge triggers huge future globals
astro0@images <- list()# SCTransform on all astrocytes together (no split/merge, no integration)
astro <- SCTransform(
astro0,
assay = "Xenium",
new.assay.name = "SCT",
verbose = FALSE
)## Log-normalised alternative
# Log-normalise, find variable features, and scale
astro_log <- NormalizeData(astro0, normalization.method = "LogNormalize", scale.factor = 10000)
astro_log <- FindVariableFeatures(astro_log, selection.method = "vst", nfeatures = 2000)
astro_log <- ScaleData(astro_log, features = VariableFeatures(astro_log))
# PCA, neighbours, clustering, UMAP
set.seed(1234)
astro_log <- RunPCA(astro_log, features = VariableFeatures(astro_log), verbose = FALSE)
astro_log <- FindNeighbors(astro_log, reduction = "pca", dims = 1:15)
astro_log <- FindClusters(astro_log, resolution = 0.3)
astro_log <- RunUMAP(astro_log, reduction = "pca", dims = 1:15, verbose = FALSE)
DimPlot(astro_log, label =T, label.size = 6, pt.size =0.9)+ NoLegend() +
ggtitle("Removed cluster 6 and 103 marker genes\nLog-normalized")
# qs_save(astro_log, "seurat_objects/20260318-astro_lognorm_0.3.qs2")DefaultAssay(astro) <- "SCT"
astro <- RunPCA(
astro,
assay = "SCT",
features = VariableFeatures(astro),
verbose = FALSE)
set.seed(1234)
# graph.name keeps the SNN graph separate from any pre-existing graphs
astro <- FindNeighbors(astro, reduction = "pca", dims = 1:30, graph.name = "astro_snn")
astro <- FindClusters(astro, graph.name = "astro_snn", resolution = 0.3)
astro <- RunUMAP(astro, reduction = "pca", dims = 1:30, verbose = FALSE)
## save cleaned object
# qs_save(astro, "seurat_objects/20260318-astro_cleaned2_0.3.qs2")## load cleaned object
astro <- qs_read("seurat_objects/20260318-astro_cleaned2_0.3.qs2")
# swap to log-normalised version when ready:
# astro <- qs_read("seurat_objects/20260318-astro_lognorm_0.3.qs2")# Elbow plot to visualise variance explained per PC
ElbowPlot(astro, ndims = 50) +
geom_vline(xintercept = 30, linetype = "dashed", color = "red") +
labs(title = "Elbow plot") +
theme_minimal()
# PCA biplots coloured by sample and treatment
p1 <- DimPlot(astro, reduction = "pca", group.by = "Slide") +
labs(title = "PCA — Slide")
p2 <- DimPlot(astro, reduction = "pca", group.by = "Treatment.Group") +
labs(title = "PCA — Treatment")
p1 + p2
resolution = 0.3

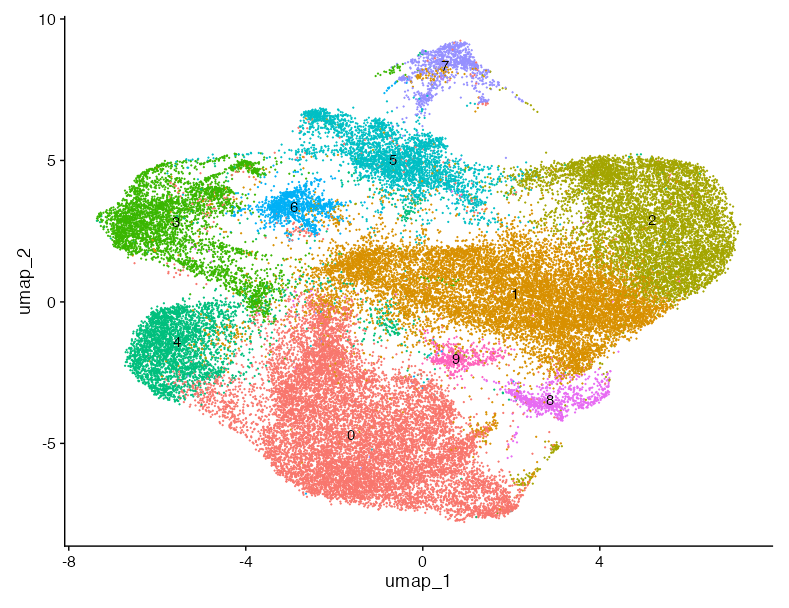
#|fig-width: 5
#|fig-height: 5
#| label: plot UMAPs 1
DimPlot(astro, label =T, label.size = 6, pt.size =0.9)+ NoLegend() +
ggtitle("Removed cluster 6 and 103 marker genes") 
DimPlot(astro, split.by = "Treatment.Group", label.size = 6, pt.size =0.9)+ NoLegend()
FindAllMArkers run on log-normalized residual expression values from SCTransform in the SCT assay (Values are continuous and typically centered around 0)
Idents(astro) <- astro$seurat_clusters
DefaultAssay(astro) <- "SCT"
all_markers <- FindAllMarkers(astro, only.pos = TRUE, densify = TRUE)
# write_csv (only marker genes removed)
# write_csv(all_markers, file = "results/20260309-astrocyte_cleaned_all_markers_0.3.csv")
#################################################################
# write_csv (cluster 6 from the original object & marker genes removed)
# write_csv(all_markers, file = "results/20260312-astrocyte_cleaned_all_markers_0.3.csv")
#################################################################
# write_csv (cluster 6 from the original object & marker genes + Akhil genes removed)
# write_csv(all_markers, file = "results/20260318-astrocyte_cleaned_all_markers_0.3.csv")
write_csv(all_markers, file = "results/20260318-astrocyte_cleaned_all_markers_0.3_logtrans.csv")source("20260217-helper_functions.R")
# Top 5 markers per cluster by avg_log2FC
top_genes <- read_csv("results/20260318-astrocyte_cleaned_all_markers_0.3.csv",
show_col_types = FALSE) |>
group_by(cluster) |>
arrange(desc(avg_log2FC), .by_group = TRUE) |>
slice_head(n = 5) |>
pull(gene) |>
unique()
fiddle(astro,
genes_of_interest = top_genes,
classification_col = "seurat_clusters",
assay = "SCT")
Based on the top 5 marker genes per cluster, we assessed whether each cluster represents true astrocytes or contaminating cell types from the initial subsetting.
FeaturePlot(astro,features = c("Aldh1l1", "Aldoc", "Gfap", "Slc7a10", "Aqp4"), ncol=2, min.cutoff = "q20")
VlnPlot(astro,features = c("Aldh1l1", "Aldoc", "Gfap", "Slc7a10", "Aqp4"),alpha = 0, ncol=2)